Research
The size and complexity of publicly available genomics data is immense, and represents the result of billions of dollars of public research expenditure. This valuable repository of data offers an unprecedented opportunity to make new discoveries relevant to basic science, as well as human health and disease. Unfortunately, the wealth of information in these datasets has gone undiscovered as most investigators can’t use it due to its scale, diverse protocols and aims, highly variable metadata, and systemic batch effects between studies.
The goal of my research is to turn these challenges into strengths – that is, to develop statistical and computational methods that leverage the scale and heterogeneity of publicly available data in order to bring the power of this data to the biomedical research community and, in so doing, maximize the new knowledge that can be obtained from this invaluable resource.
Identifying spatially varying correlation between genes in spatial transcriptomics data
Recent advances in spatially resolved transcriptomics technologies enable both the measurement of genome-wide gene expression profiles and their mapping to spatial locations within a tissue. We present SpatialCorr, a method for identifying sets of genes with spatially varying correlation structure. Given a collection of gene sets pre-defined by a user, SpatialCorr tests for spatially induced differences in the correlation of each gene set within tissue regions, as well as between and among regions.
- Bernstein, M.N., Ni, Z., Prasad, A., Brown, J., Mohanty, C., Stewart, R., Newton, M.A., and Kendziorski, C. (2022). SpatialCorr: Identifying gene sets with spatially varying correlation structure. Cell Reports Methods, 2(12), 100369.

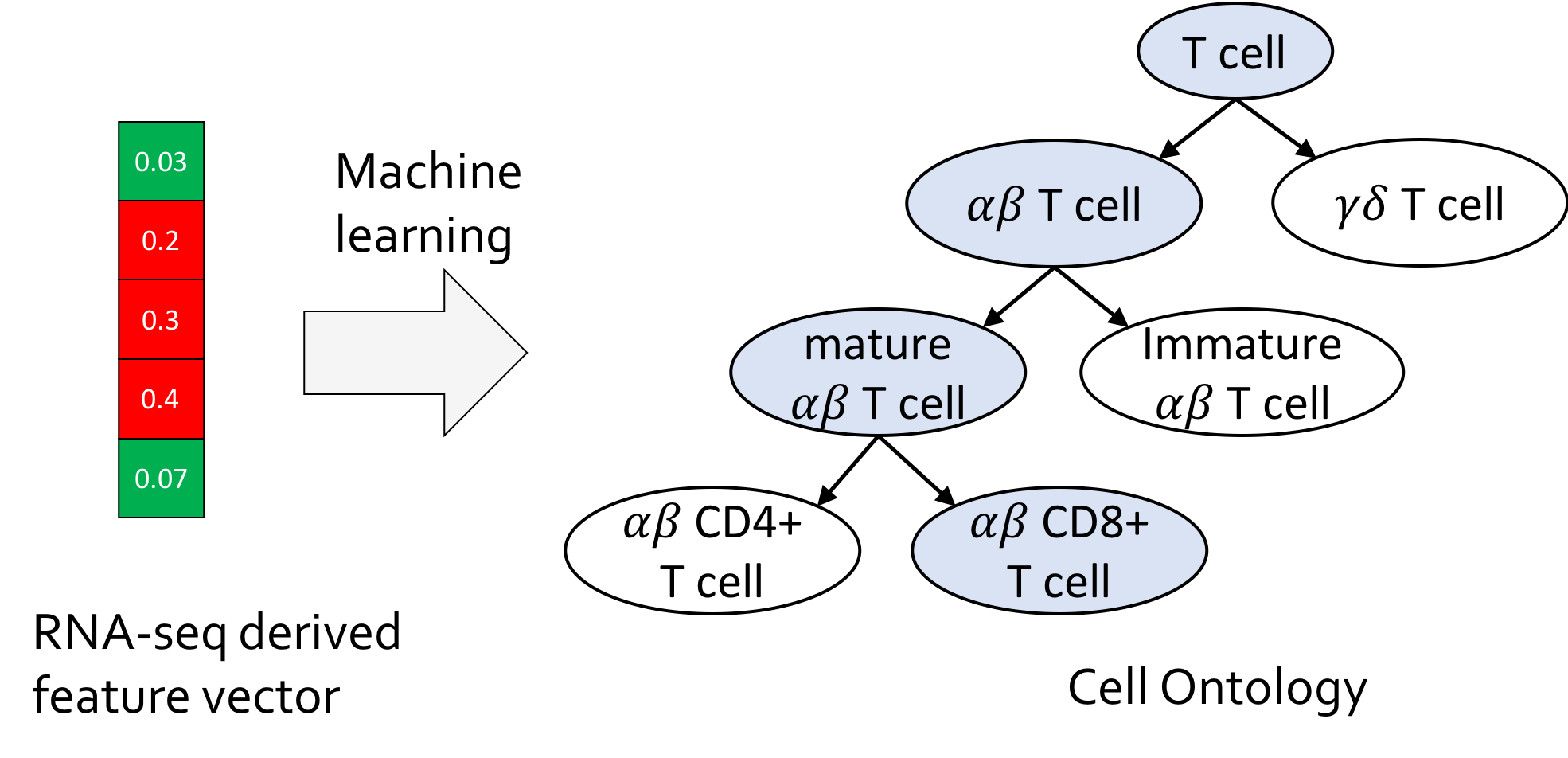
Cell type classification with the Cell Ontology
Cell type annotation is a fundamental task in the analysis of single-cell RNA-sequencing data. We present CellO, a machine learning-based tool for annotating human RNA-seq data with the Cell Ontology. CellO enables accurate and standardized cell type classification by considering the rich hierarchical structure of known cell types. Furthemore, CellO comes pre-trained on a novel, comprehensive dataset of human, healthy, untreated primary samples in the Sequence Read Archive (SRA) which, to the best of our knowledge, is the most diverse curated collection of primary cell data to date.
- Bernstein, M.N., Ma, J., Gleicher, M., and Dewey, C.N. (2021). CellO: Comprehensive and hierarchical cell type classification of human cells with the Cell Ontology. iScience. 24(1), 101913.
- Bernstein, M.N., Dewey, C.N. (2021). Annotating cell types in human single-cell RNA-seq data with CellO. STAR Protocols, 2(3), 100705.
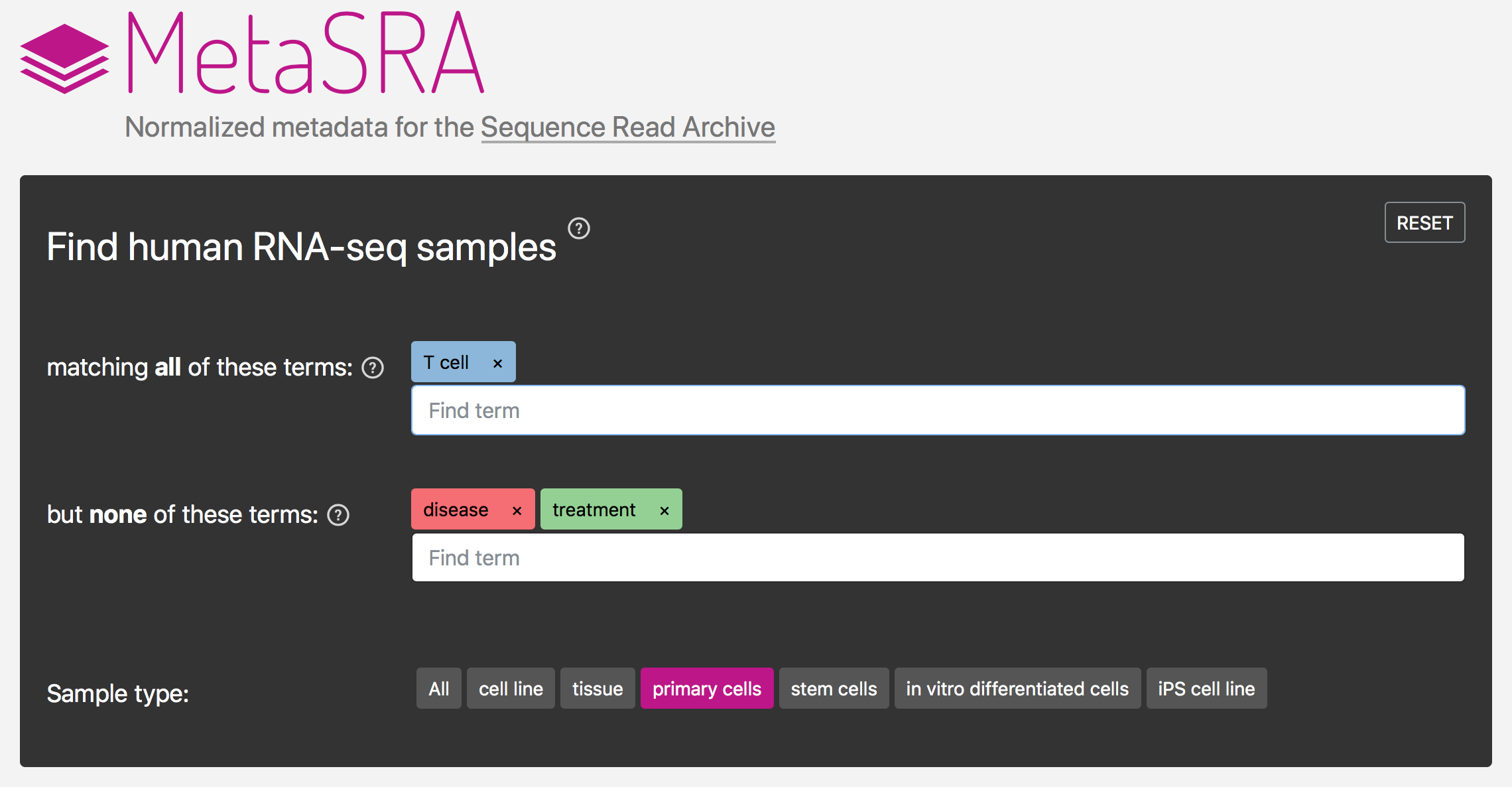
Standardizing metadata for large, public genomics databases
The NCBI’s Sequence Read Archive (SRA) promises great biological insight if one could analyze the data in the aggregate; however, the data remain largely underutilized, in part, due to the poor structure of the metadata associated with each sample. We developed the MetaSRA, a novel computational pipeline and associated database for standardizing the metadata associated with samples in the SRA by mapping each sample to biomedical ontologies. See my talk describing the MetaSRA from ISMB 2018.
- Bernstein, M.N., Doan, A., and Dewey, C.N. (2017). MetaSRA: Normalized human sample-specific metadata for the Sequence Read Archive. Bioinformatics. 33(18), 2914–2923.
- Bernstein, M.N., et al. (2020). Jupyter notebook-based tools for building structured datasets from the Sequence Read Archive. F1000 Research. 9(376).
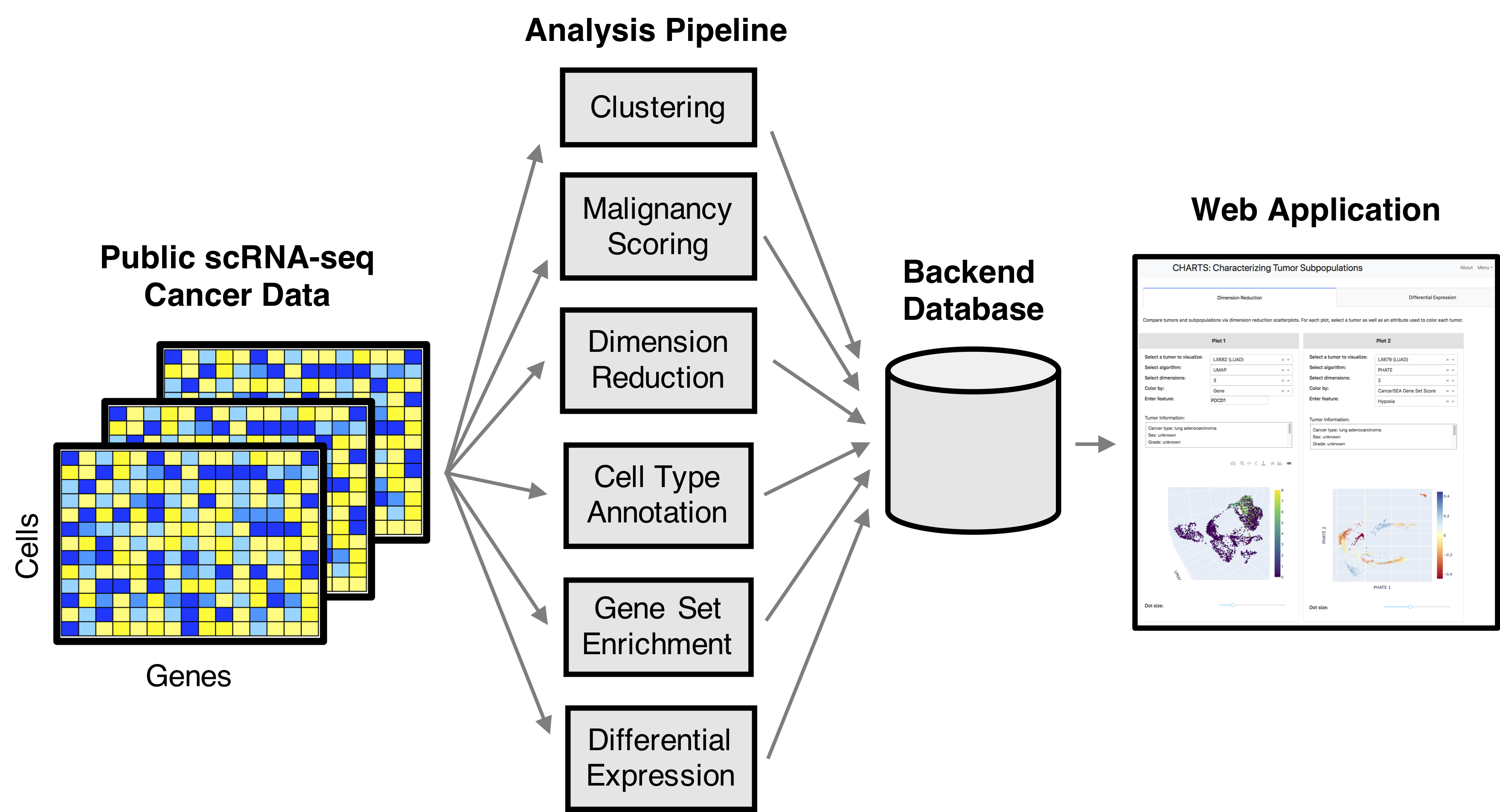
Webtools for exploring public single-cell cancer data
Single-cell RNA-seq (scRNA-seq) enables the profiling of genome-wide gene expression at the single-cell level and in so doing facilitates insight into and information about cellular heterogeneity within a tissue. This is especially important in cancer, where tumor and tumor microenvironment heterogeneity directly impact development, maintenance, and progression of disease. We present CHARacterizing Tumor Subpopulations (CHARTS), a computational pipeline and web application for analyzing, characterizing, and integrating publicly available scRNA-seq cancer datasets. CHARTS is freely available at charts.morgridge.org.
- Bernstein, M.N., Ni, Z., Collins, M., Burkard, M.E., Kendziorski, C., and Stewart, R. (2021). CHARTS: A web application for characterizing and comparing tumor subpopulations in publicly available single-cell RNA-seq datasets. BMC Bioinformatics. 22(83).