
Projects
Here are some of the research projects I have worked on over the years.
Monkeybread: Exploring cellular niches in spatial transcriptomics data
Monkeybread is a Python toolkit that provides broad functionality for analyzing and visualizing cellular organization, cellular niches, and intercellular communication via ligand-receptor interactions. Monkeybread provides functions for visualization methods and lightweight statistical methods for exploring spatial transcriptomics data.
- GitHub: https://github.com/immunitastx/monkeybread
- Documentation: https://monkeybread.readthedocs.io/en/latest/
- Paper: https://doi.org/10.1101/2023.09.14.557736
SpatialCorr: A statistical method for identifying spatially varying correlation
Recent advances in spatially resolved transcriptomics technologies enable both the measurement of genome-wide gene expression profiles and their mapping to spatial locations within a tissue. SpatialCorr is a method for identifying sets of genes with spatially varying correlation structure. Given a collection of gene sets pre-defined by a user, SpatialCorr tests for spatially induced differences in the correlation of each gene set within tissue regions, as well as between and among regions.
- GitHub: https://github.com/mbernste/SpatialCorr
- Documentation: https://spatialcorr.readthedocs.io/en/latest/index.html
- Paper: https://doi.org/10.1016/j.crmeth.2022.100369

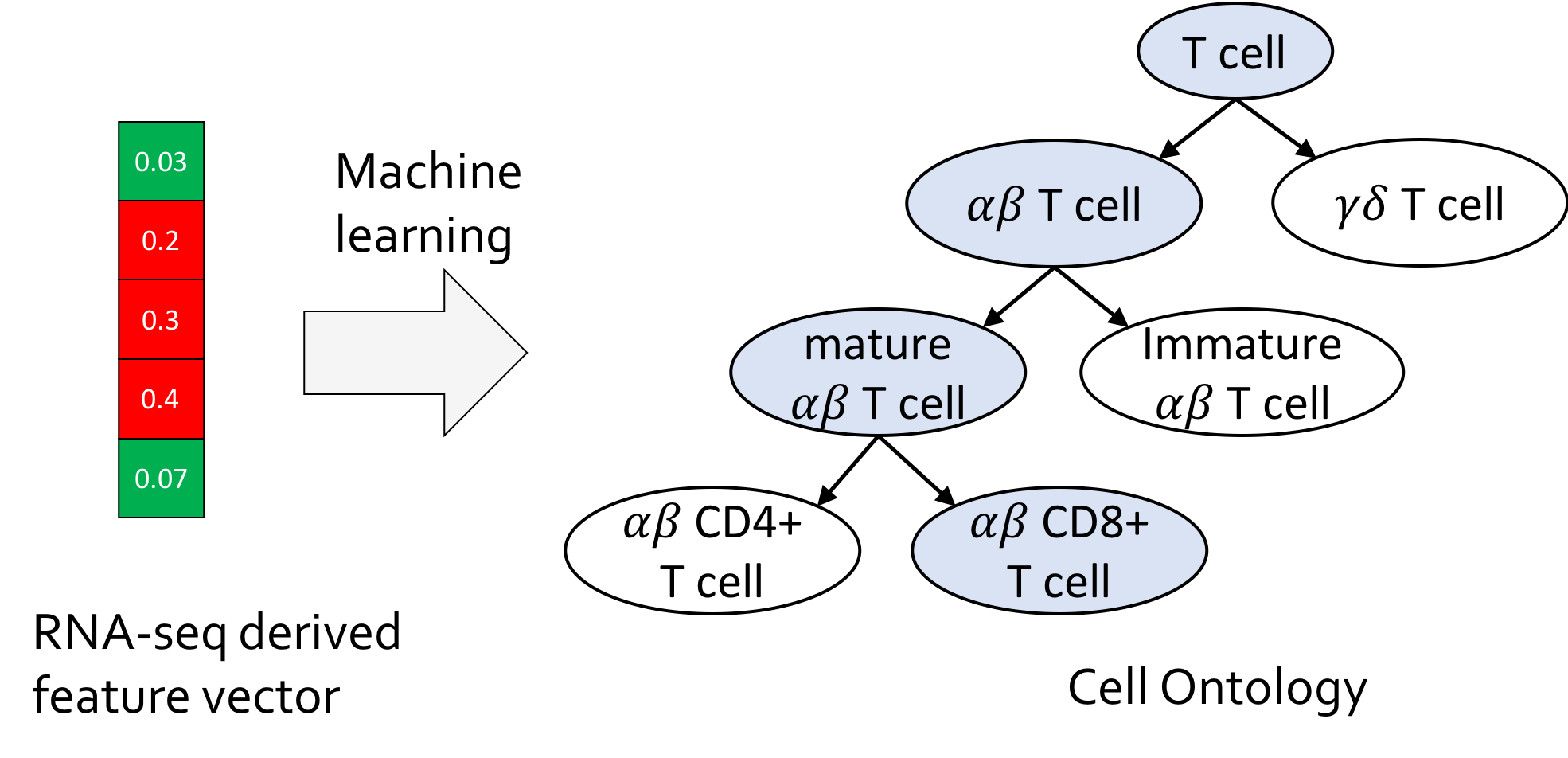
CellO: Hierarchical cell type classification against the Cell Ontology
Cell type annotation is a fundamental task in the analysis of single-cell RNA-sequencing data. We present CellO, a machine learning-based tool for annotating human RNA-seq data with the Cell Ontology. CellO enables accurate and standardized cell type classification by considering the rich hierarchical structure of known cell types. Furthemore, CellO comes pre-trained on a novel, comprehensive dataset of human, healthy, untreated primary samples in the Sequence Read Archive (SRA) which, to the best of our knowledge, is the most diverse curated collection of primary cell data to date.
- GitHub: https://github.com/deweylab/CellO
- Paper: https://doi.org/10.1016/j.isci.2020.101913
- Website: https://uwgraphics.github.io/CellOViewer/
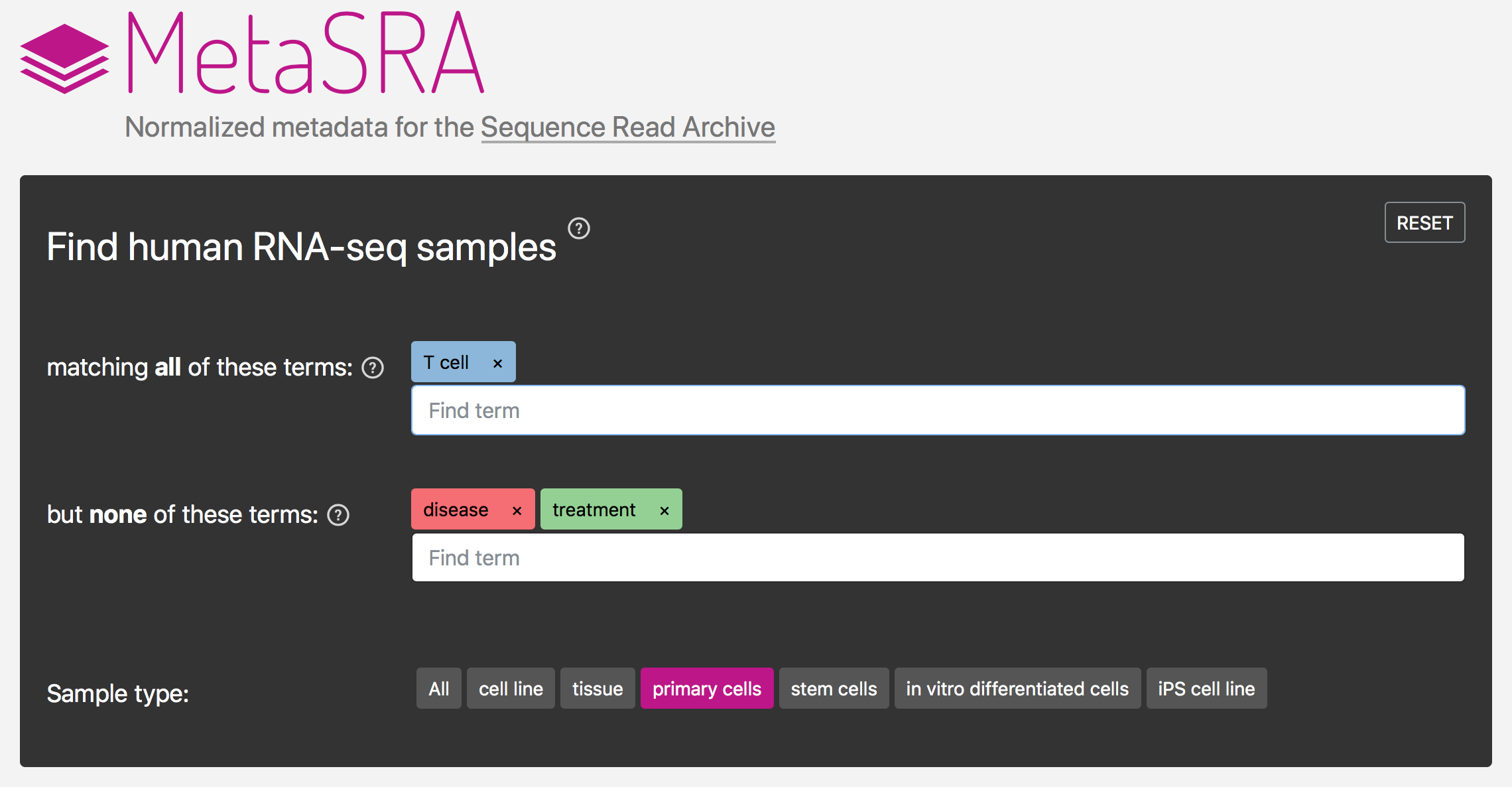
MetaSRA: Standardized metadata for the Sequence Read Archive
The NCBI’s Sequence Read Archive (SRA) promises great biological insight if one could analyze the data in the aggregate; however, the data remain largely underutilized, in part, due to the poor structure of the metadata associated with each sample. We developed the MetaSRA, a novel computational pipeline and associated database for standardizing the metadata associated with samples in the SRA by mapping each sample to biomedical ontologies. See describing the MetaSRA from ISMB 2018.
- Website: http://metasra.biostat.wisc.edu
- GitHub:
- Paper: https://doi.org/10.1093/bioinformatics/btx334
- Talk: At ISMB 2018
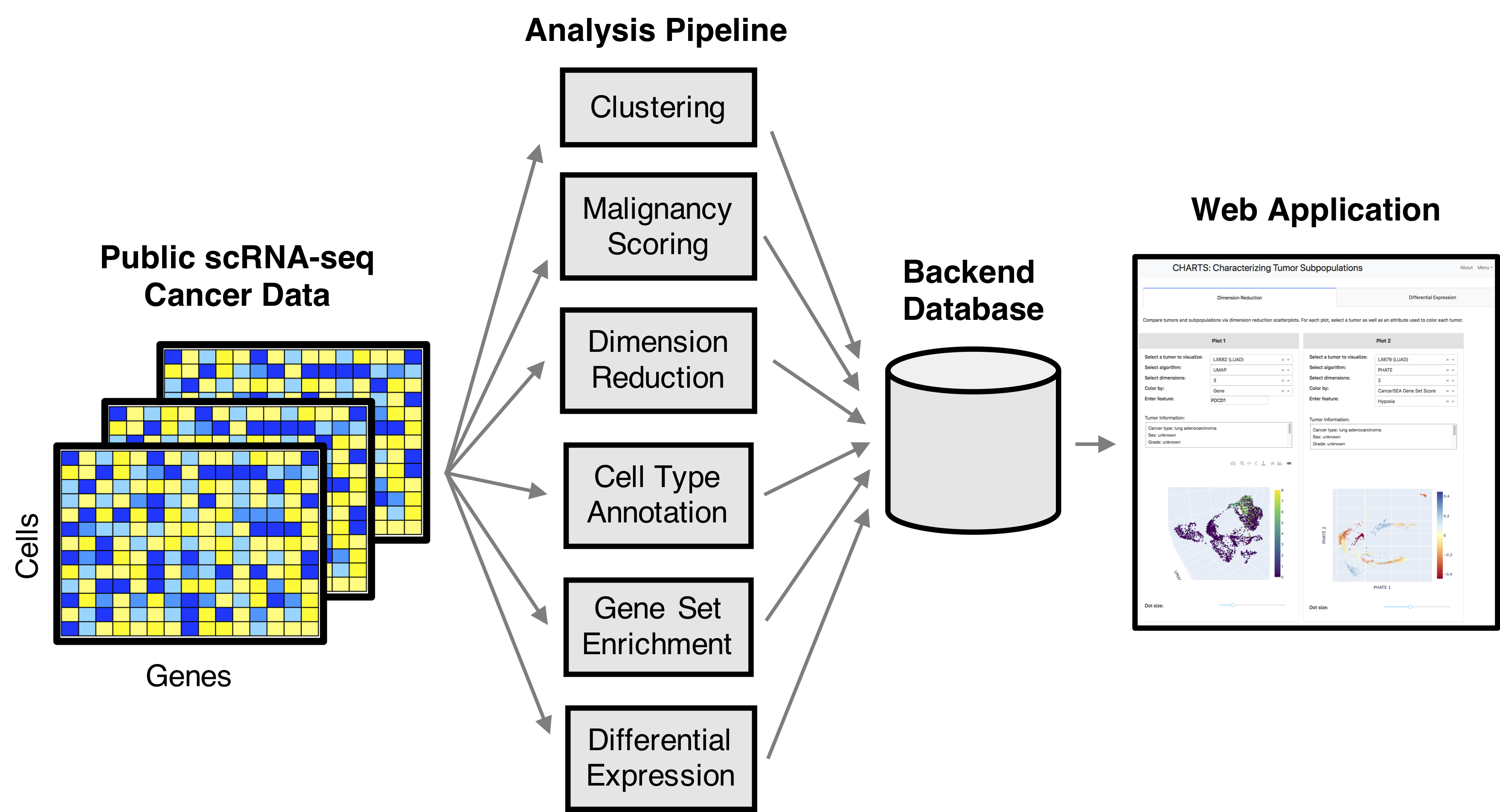
CHARTS: A web tool for exploring single-cell cancer data
Single-cell RNA-seq (scRNA-seq) enables the profiling of genome-wide gene expression at the single-cell level and in so doing facilitates insight into and information about cellular heterogeneity within a tissue. This is especially important in cancer, where tumor and tumor microenvironment heterogeneity directly impact development, maintenance, and progression of disease. CHARacterizing Tumor Subpopulations (CHARTS) is a computational pipeline and web application for analyzing, characterizing, and integrating publicly available scRNA-seq cancer datasets. CHARTS is freely available at charts.morgridge.org.
- Website: https://charts.morgridge.org
- GitHub: https://github.com/stewart-lab/CHARTS
- Paper: https://doi.org/10.1186/s12859-021-04021-x
Build structured datasets from the Sequence Read Archive
Recently, the MetaSRA project standardized these metadata by annotating each sample with terms from biomedical ontologies. In this work, we present a pair of Jupyter notebook-based tools that utilize the MetaSRA for building structured datasets from the SRA in order to facilitate secondary analyses of the SRA’s human RNA-seq data. The first tool, called the Case-Control Finder, finds suitable case and control samples for a given disease or condition where the cases and controls are matched by tissue or cell type. The second tool, called the Series Finder, finds ordered sets of samples for the purpose of addressing biological questions pertaining to changes over a numerical property such as time. These tools were the result of a three-day-long NCBI-sponsored hackathon in March 2019 held at the University of North Carolina at Chapel Hill.